STARmap Mouse Cortex¶
Warning
This tutorial was written with Giotto version 0.3.6.9046, your version is 1.0.4. This is a more recent version and results should be reproducible.
Install Giotto and Python Modules¶
To run this vignette you need to install all of the necessary Python modules.
Important
Python module installation can be done either automatically via our installation tool (from within R) (see step 2.2A) or manually (see step 2.2B).
See Part 2.2 Giotto-Specific Python Packages of our Giotto Installation section for step-by-step instructions.
Set-Up Giotto¶
library(Giotto)
Set A Working Directory¶
#results_folder = '/path/to/directory/'
results_folder = '/Volumes/Ruben_Seagate/Dropbox (Personal)/Projects/GC_lab/Ruben_Dries/190225_spatial_package/Results/Visium/Brain/201226_results//'
Set A Giotto Python Path¶
# set python path to your preferred python version path
# set python path to NULL if you want to automatically install (only the 1st time) and use the giotto miniconda environment
python_path = NULL
if(is.null(python_path)) {
installGiottoEnvironment()
}
Data Explanation¶
Wang et al. created a 3D spatial expression dataset consisting of 28 genes from 32,845 single cells in a visual cortex volume using the STARmap technology.
The STARmap data to run this tutorial can be found here. Alternatively you can use the getSpatialDataset to automatically download this dataset like we do in this example.
Download Datasest¶
# download data to working directory
# if wget is installed, set method = 'wget'
# if you run into authentication issues with wget, then add " extra = '--no-check-certificate' "
getSpatialDataset(dataset = 'starmap_3D_cortex', directory = results_folder, method = 'wget')
1. Giotto Global Instructions and Preparations¶
## instructions allow us to automatically save all plots into a chosen results folder
instrs = createGiottoInstructions(show_plot = FALSE,
save_plot = TRUE,
save_dir = results_folder,
python_path = python_path)
expr_path = paste0(results_folder, "STARmap_3D_data_expression.txt")
loc_path = paste0(results_folder, "STARmap_3D_data_cell_locations.txt")
2. Create Giotto Object and Process Data¶
## create
STAR_test <- createGiottoObject(raw_exprs = expr_path,
spatial_locs = loc_path,
instructions = instrs)
## filter raw data
# pre-test filter parameters
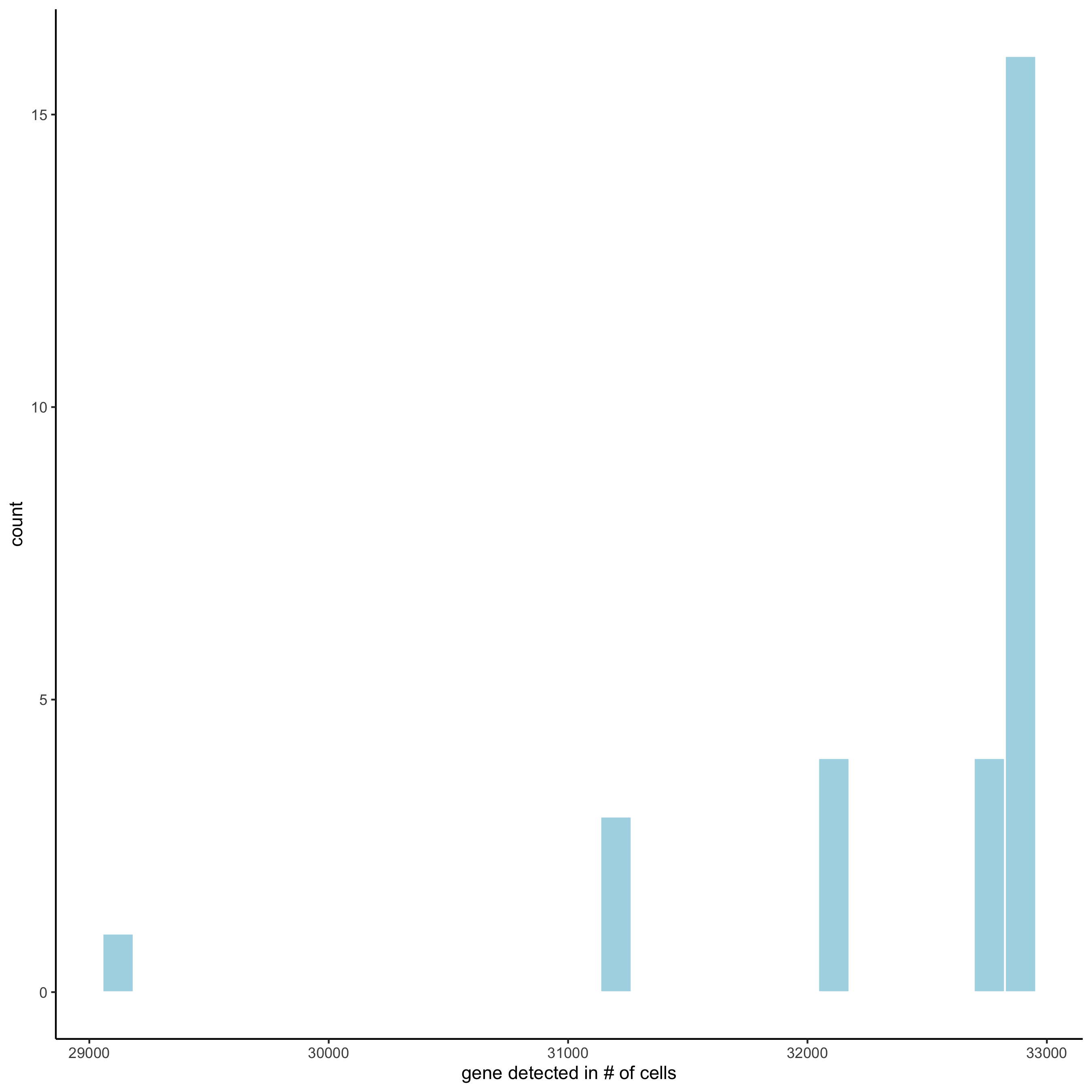
filterDistributions(STAR_test, detection = 'genes',
save_param = list(save_name = '2_a_distribution_genes'))
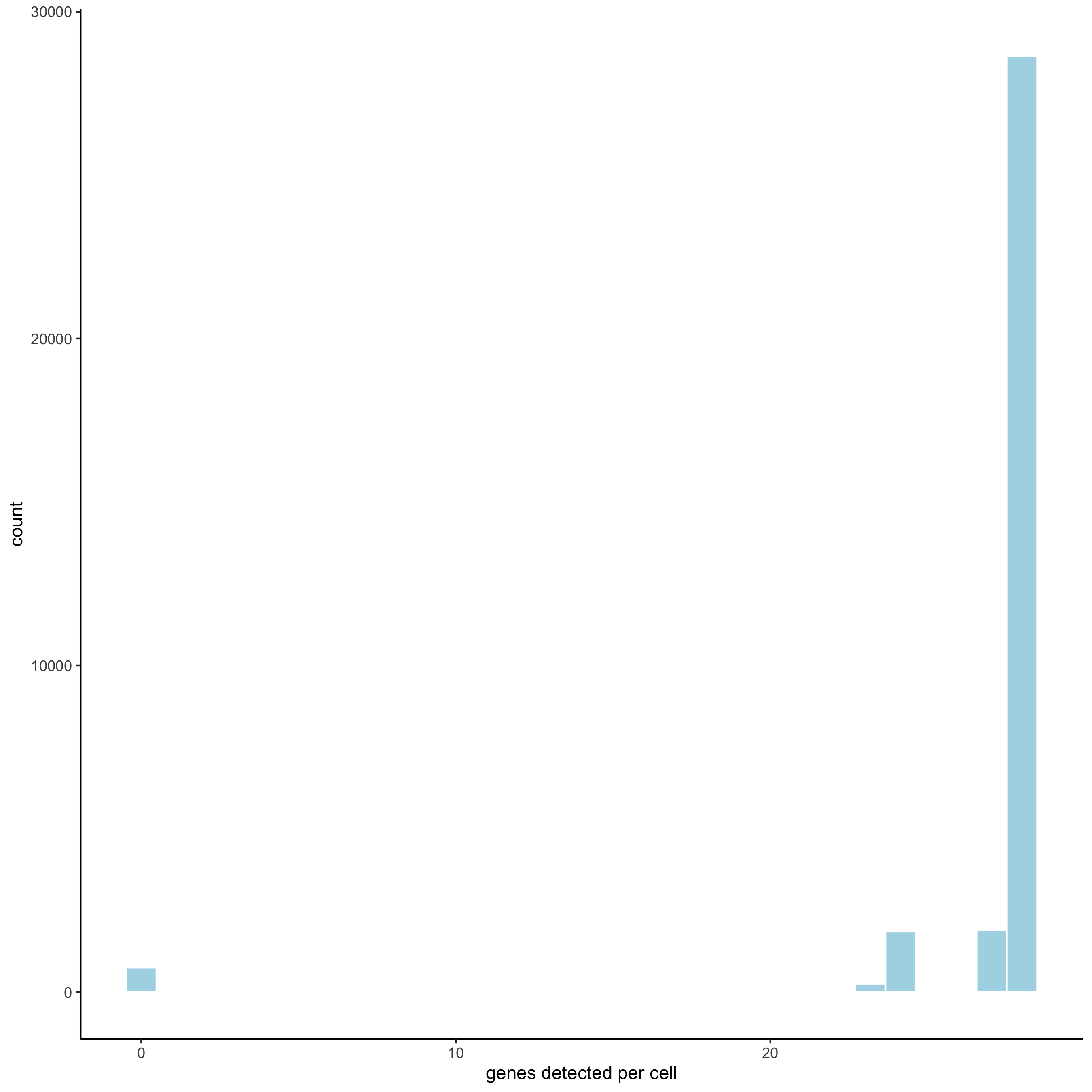
filterDistributions(STAR_test, detection = 'cells',
save_param = list(save_name = '2_b_distribution_cells'))
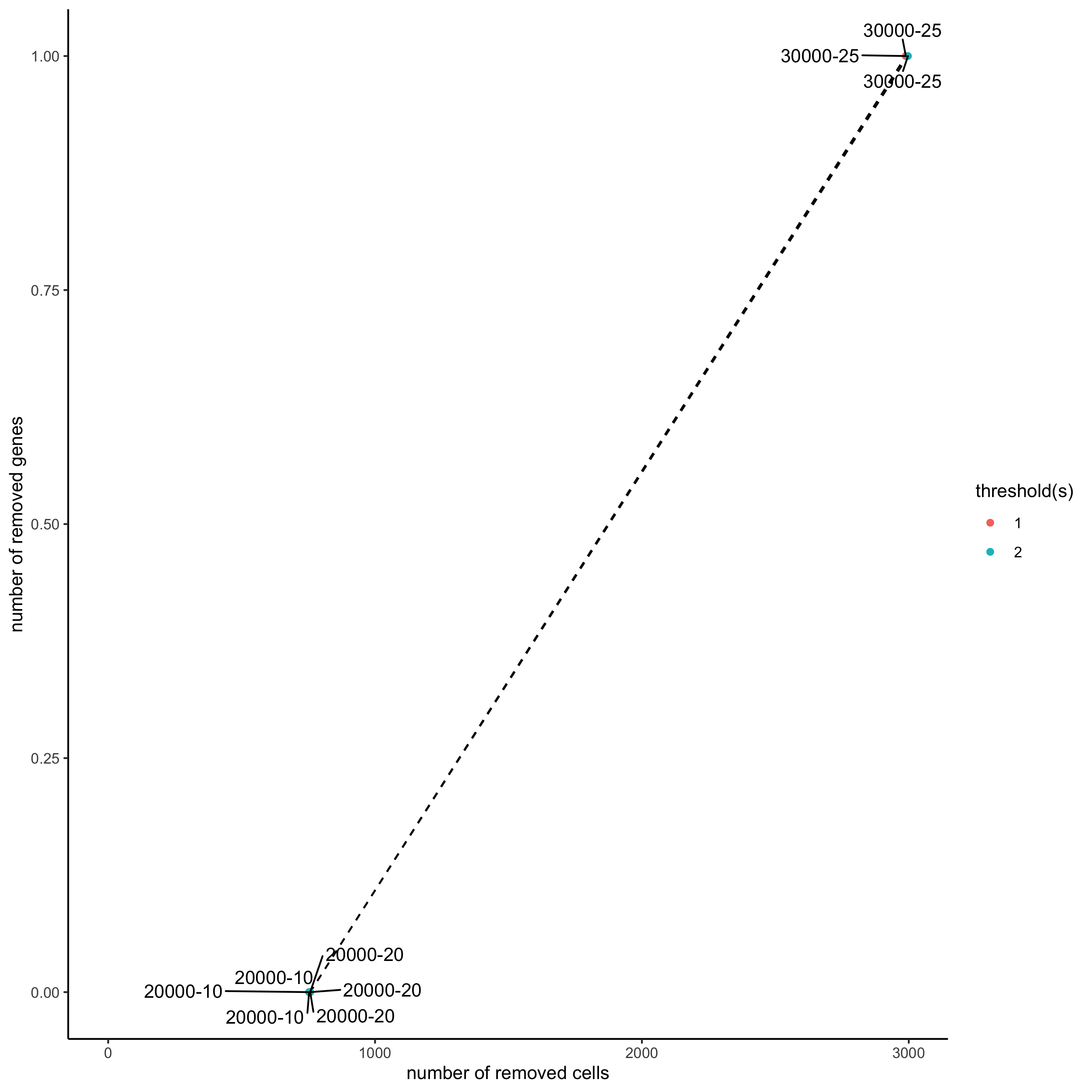
filterCombinations(STAR_test, expression_thresholds = c(1, 1,2),
gene_det_in_min_cells = c(20000, 20000, 30000),
min_det_genes_per_cell = c(10, 20, 25),
save_param = list(save_name = '2_c_distribution_filters'))
# filter
STAR_test <- filterGiotto(gobject = STAR_test,
gene_det_in_min_cells = 20000,
min_det_genes_per_cell = 20)
## normalize
STAR_test <- normalizeGiotto(gobject = STAR_test, scalefactor = 10000, verbose = T)
STAR_test <- addStatistics(gobject = STAR_test)
STAR_test <- adjustGiottoMatrix(gobject = STAR_test, expression_values = c('normalized'),
batch_columns = NULL, covariate_columns = c('nr_genes', 'total_expr'),
return_gobject = TRUE,
update_slot = c('custom'))
## visualize
# 3D
spatPlot3D(gobject = STAR_test, point_size = 2,
save_param = list(save_name = '2_d_spatplot_3D'))
3. Dimension Reduction¶
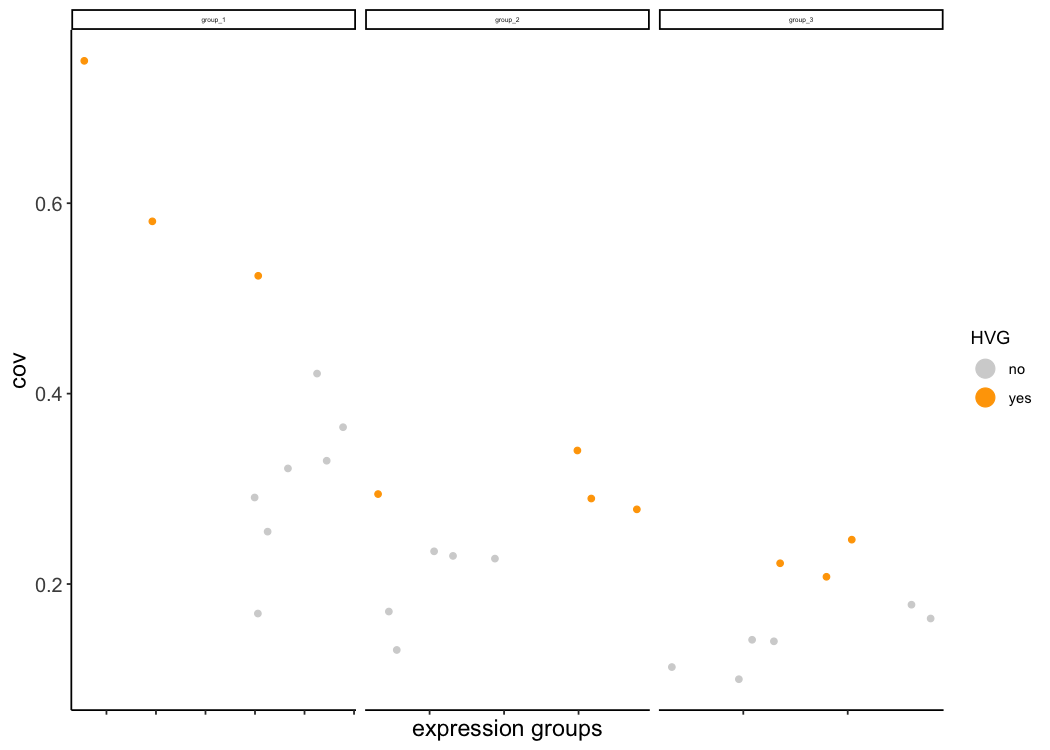
STAR_test <- calculateHVG(gobject = STAR_test, method = 'cov_groups',
zscore_threshold = 0.5, nr_expression_groups = 3,
save_param = list(save_name = '3_a_HVGplot', base_height = 5, base_width = 5))
# too few highly variable genes
# genes_to_use = NULL is the default and will use all genes available
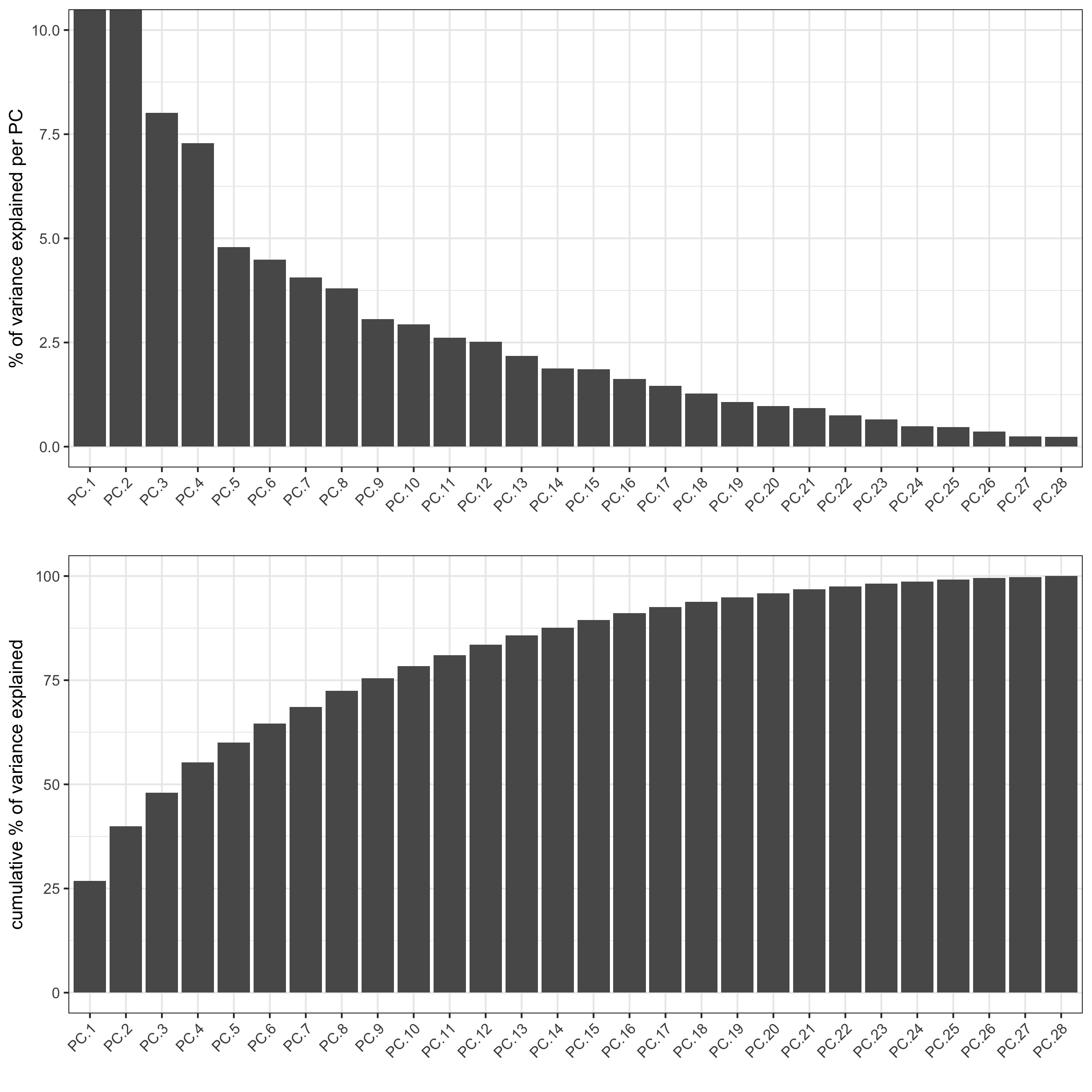
STAR_test <- runPCA(gobject = STAR_test, genes_to_use = NULL, scale_unit = F,method = 'factominer')
signPCA(STAR_test,
save_param = list(save_name = '3_b_signPCs'))
STAR_test <- runUMAP(STAR_test, dimensions_to_use = 1:8, n_components = 3, n_threads = 4)
plotUMAP_3D(gobject = STAR_test,
save_param = list(save_name = '3_c_UMAP'))
4. Clustering¶
## sNN network (default)
STAR_test <- createNearestNetwork(gobject = STAR_test, dimensions_to_use = 1:8, k = 15)
## Leiden clustering
STAR_test <- doLeidenCluster(gobject = STAR_test, resolution = 0.2, n_iterations = 100,
name = 'leiden_0.2')
plotUMAP_3D(gobject = STAR_test, cell_color = 'leiden_0.2',show_center_label = F,
save_param = list(save_name = '4_a_UMAP'))
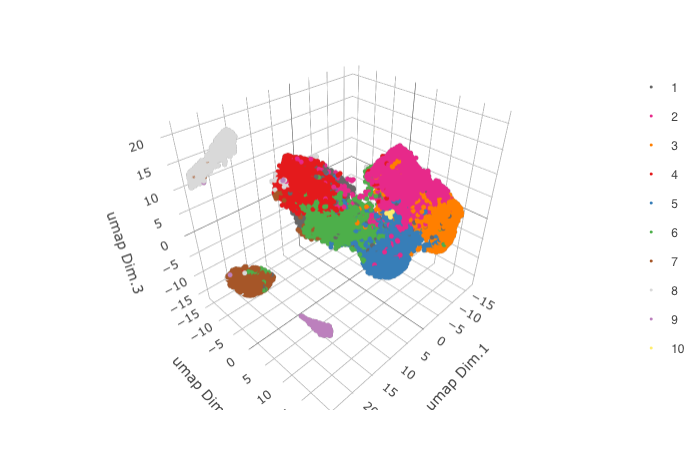
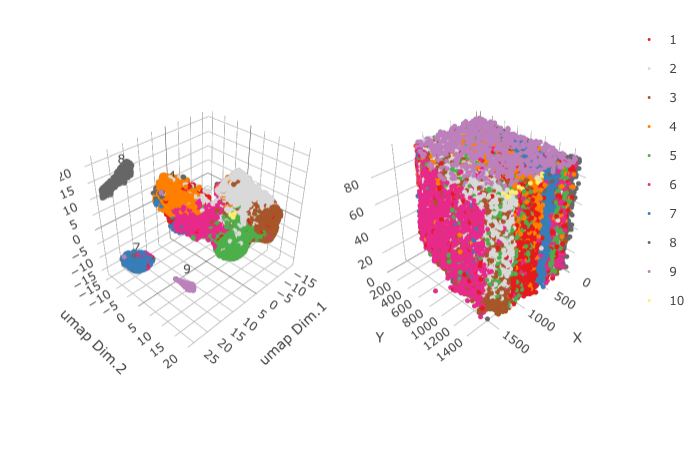
5. Co-Visualization¶
spatDimPlot3D(gobject = STAR_test,
cell_color = 'leiden_0.2',
save_param = list(save_name = '5_a_spatDimPlot'))
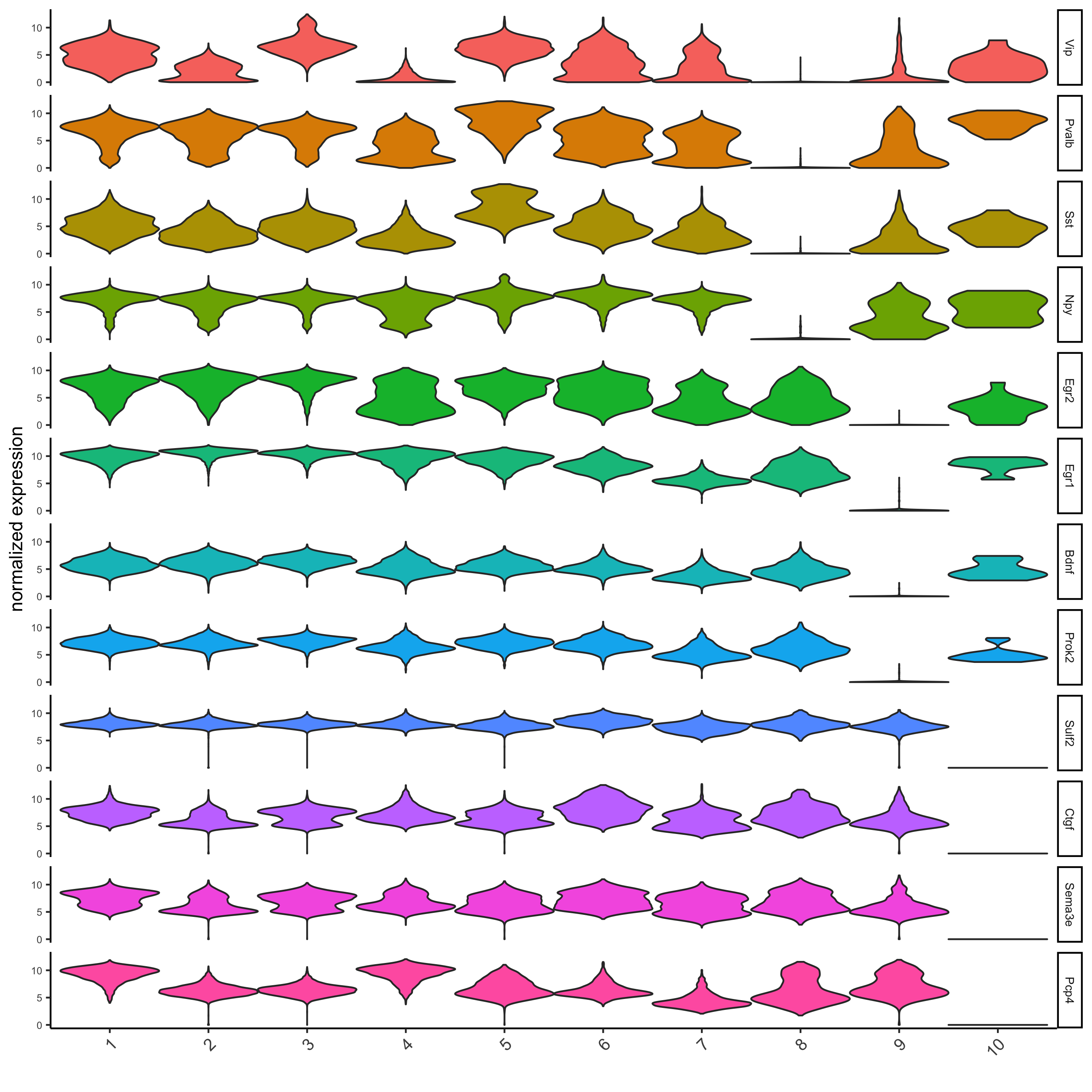
6. Differential Expression¶
markers = findMarkers_one_vs_all(gobject = STAR_test,
method = 'gini',
expression_values = 'normalized',
cluster_column = 'leiden_0.2',
min_expr_gini_score = 2,
min_det_gini_score = 2,
min_genes = 5, rank_score = 2)
markers[, head(.SD, 2), by = 'cluster']
# violinplot
violinPlot(STAR_test, genes = unique(markers$genes), cluster_column = 'leiden_0.2',
strip_position = "right", save_param = list(save_name = '6_a_violinplot'))
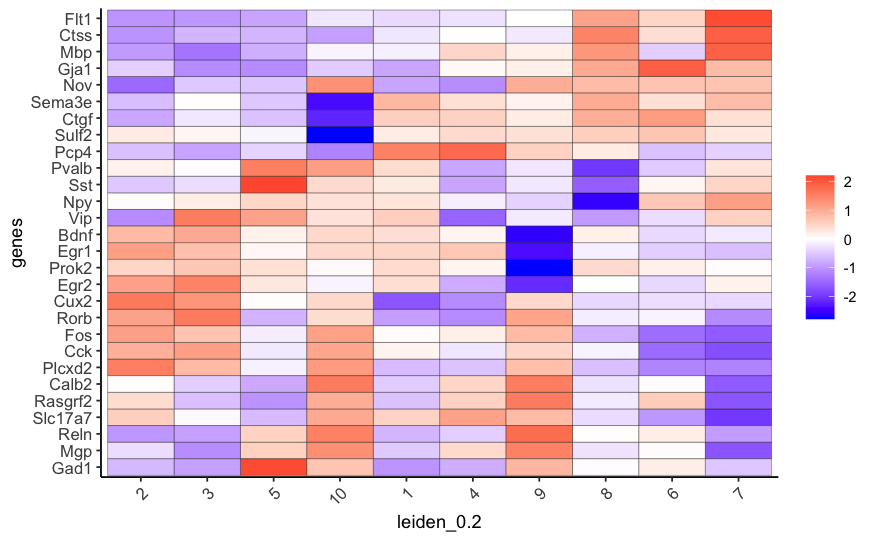
# cluster heatmap
plotMetaDataHeatmap(STAR_test, expression_values = 'scaled',
metadata_cols = c('leiden_0.2'),
save_param = list(save_name = '6_b_metaheatmap'))
7. Cell-Type Annotation¶
## general cell types
clusters_cell_types_cortex = c('excit','excit','excit', 'inh', 'excit',
'other', 'other', 'other', 'inh', 'inh')
names(clusters_cell_types_cortex) = c(1:10)
STAR_test = annotateGiotto(gobject = STAR_test, annotation_vector = clusters_cell_types_cortex,
cluster_column = 'leiden_0.2', name = 'general_cell_types')
plotMetaDataHeatmap(STAR_test, expression_values = 'scaled',
metadata_cols = c('general_cell_types'),
save_param = list(save_name = '7_a_metaheatmap'))

## detailed cell types
clusters_cell_types_cortex = c('L5','L4','L2/3', 'PV', 'L6',
'Astro', 'Olig1', 'Olig2', 'Calretinin', 'SST')
names(clusters_cell_types_cortex) = c(1:10)
STAR_test = annotateGiotto(gobject = STAR_test, annotation_vector = clusters_cell_types_cortex,
cluster_column = 'leiden_0.2', name = 'cell_types')
plotUMAP_3D(STAR_test, cell_color = 'cell_types', point_size = 1.5,show_center_label = F,
save_param = list(save_name = '7_b_UMAP'))

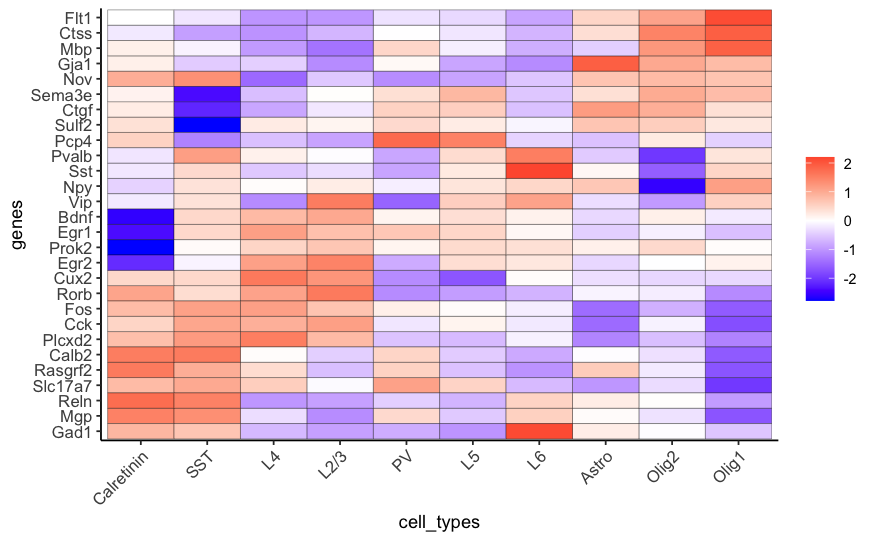
plotMetaDataHeatmap(STAR_test, expression_values = 'scaled',
metadata_cols = c('cell_types'),
custom_cluster_order = c("Calretinin", "SST", "L4", "L2/3", "PV", "L5", "L6", "Astro", "Olig2", "Olig1"),
save_param = list(save_name = '7_c_metaheatmap'))
8. Cell Type Co-Visualization¶
# create consistent color code
mynames = unique(pDataDT(STAR_test)$cell_types)
mycolorcode = Giotto:::getDistinctColors(n = length(mynames))
names(mycolorcode) = mynames
spatDimPlot3D(gobject = STAR_test,
cell_color = 'cell_types',show_center_label = F,
save_param = list(save_name = '8_a_spatdimplot'))

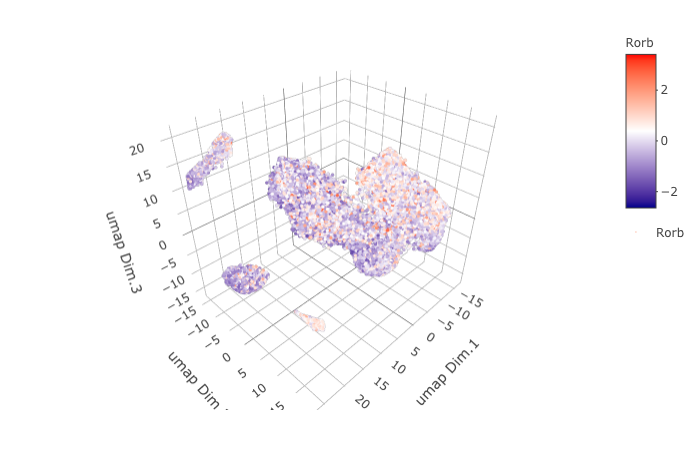
9. Gene Expression Visualization¶
dimGenePlot3D(STAR_test, expression_values = 'scaled',
genes = "Rorb",
genes_high_color = 'red', genes_mid_color = 'white', genes_low_color = 'darkblue',
save_param = list(save_name = '9_a_dimGenePlot'))
spatGenePlot3D(STAR_test,
expression_values = 'scaled',
genes = "Rorb",
show_other_cells = F,
genes_high_color = 'red', genes_mid_color = 'white', genes_low_color = 'darkblue',
save_param = list(save_name = '9_b_spatGenePlot'))
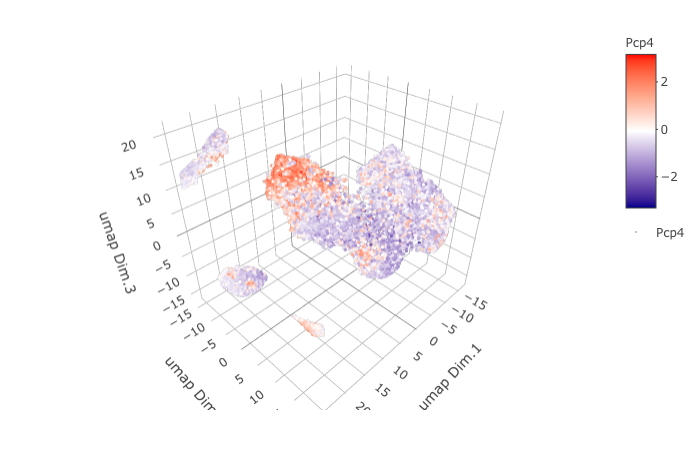
dimGenePlot3D(STAR_test, expression_values = 'scaled',
genes = "Pcp4",
genes_high_color = 'red', genes_mid_color = 'white', genes_low_color = 'darkblue',
save_param = list(save_name = '9_c_dimGenePlot'))
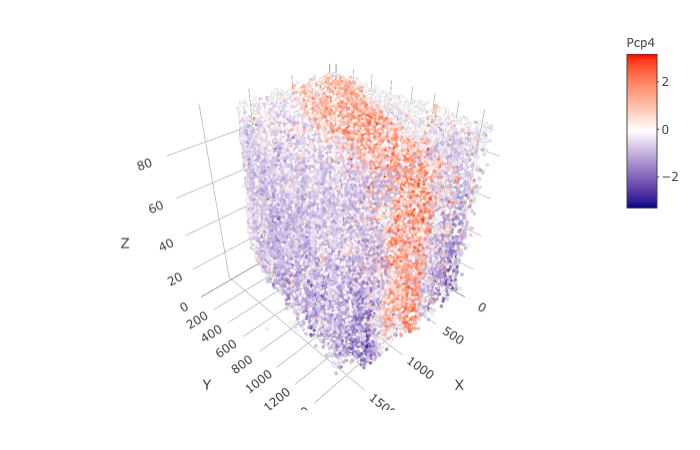
spatGenePlot3D(STAR_test,
expression_values = 'scaled',
genes = "Pcp4",
show_other_cells = F,
genes_high_color = 'red', genes_mid_color = 'white', genes_low_color = 'darkblue',
save_param = list(save_name = '9_d_spatGenePlot'))
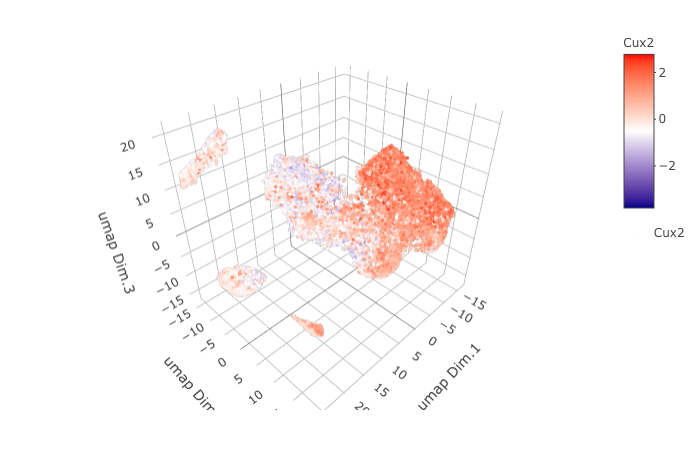
dimGenePlot3D(STAR_test, expression_values = 'scaled',
genes = "Cux2",
genes_high_color = 'red', genes_mid_color = 'white', genes_low_color = 'darkblue',
save_param = list(save_name = '9_e_dimGenePlot'))

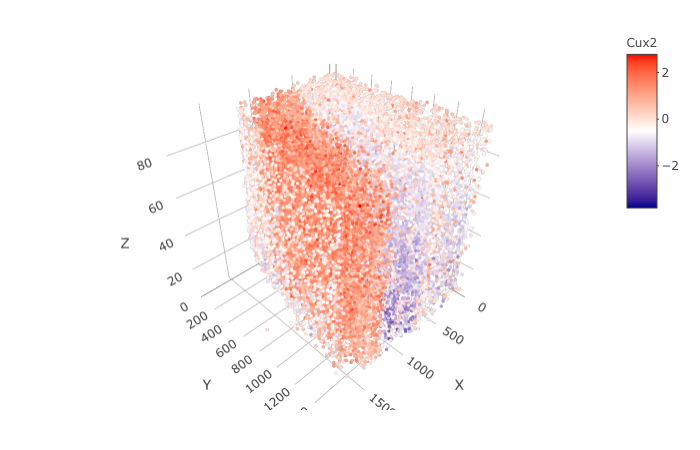
spatGenePlot3D(STAR_test,
expression_values = 'scaled',
genes = "Cux2",
show_other_cells = F,
genes_high_color = 'red', genes_mid_color = 'white', genes_low_color = 'darkblue',
save_param = list(save_name = '9_f_spatGenePlot'))
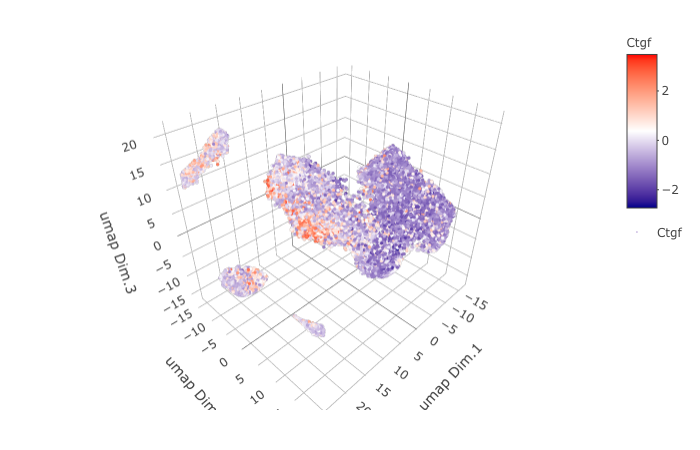
dimGenePlot3D(STAR_test, expression_values = 'scaled',
genes = "Ctgf",
genes_high_color = 'red', genes_mid_color = 'white', genes_low_color = 'darkblue',
save_param = list(save_name = '9_g_dimGenePlot'))
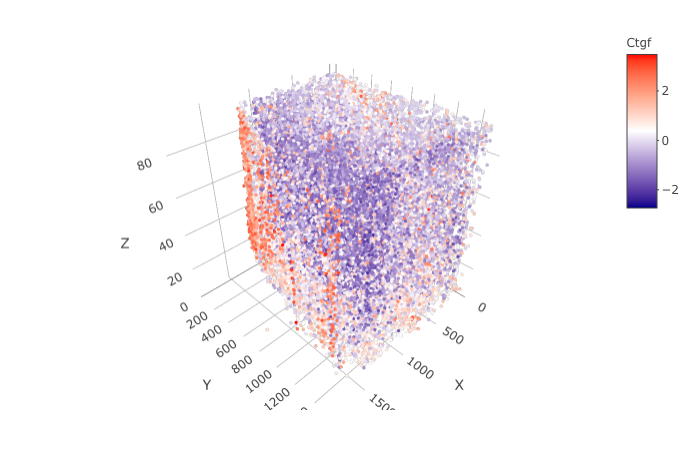
spatGenePlot3D(STAR_test,
expression_values = 'scaled',
genes = "Ctgf",
show_other_cells = F,
genes_high_color = 'red', genes_mid_color = 'white', genes_low_color = 'darkblue',
save_param = list(save_name = '9_h_spatGenePlot'))
10. Virtual Cross Section¶
STAR_test <- createSpatialNetwork(gobject = STAR_test, delaunay_method = 'delaunayn_geometry')
STAR_test = createCrossSection(STAR_test,method="equation",
equation=c(0,1,0,600),
extend_ratio = 0.6)
insertCrossSectionSpatPlot3D(STAR_test, cell_color = 'cell_types', axis_scale = 'cube',
point_size = 2,
cell_color_code = mycolorcode)

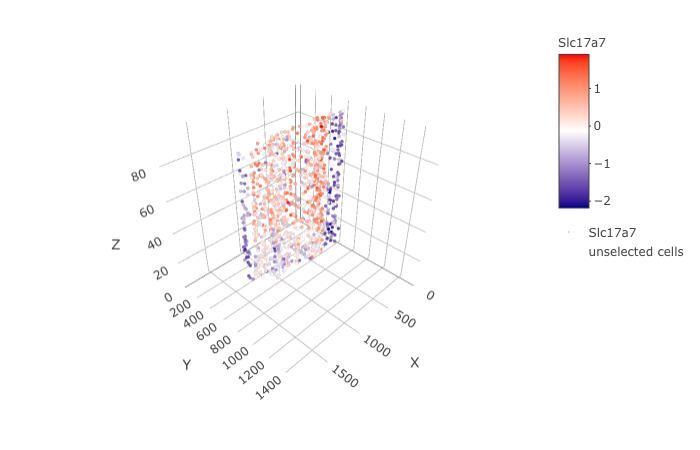
insertCrossSectionGenePlot3D(STAR_test, expression_values = 'scaled', axis_scale = "cube",
genes = "Slc17a7")
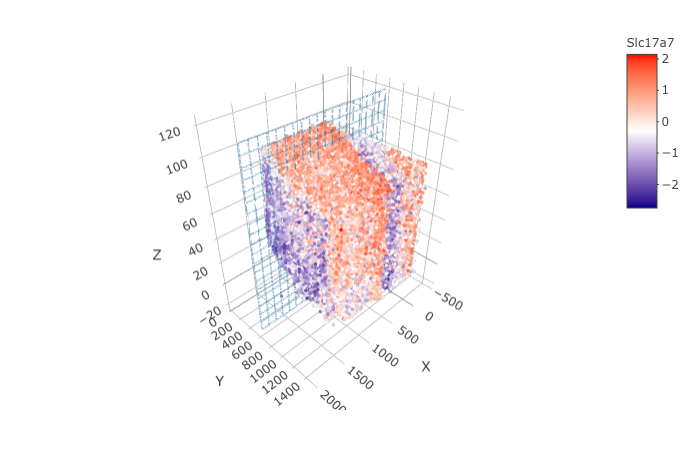
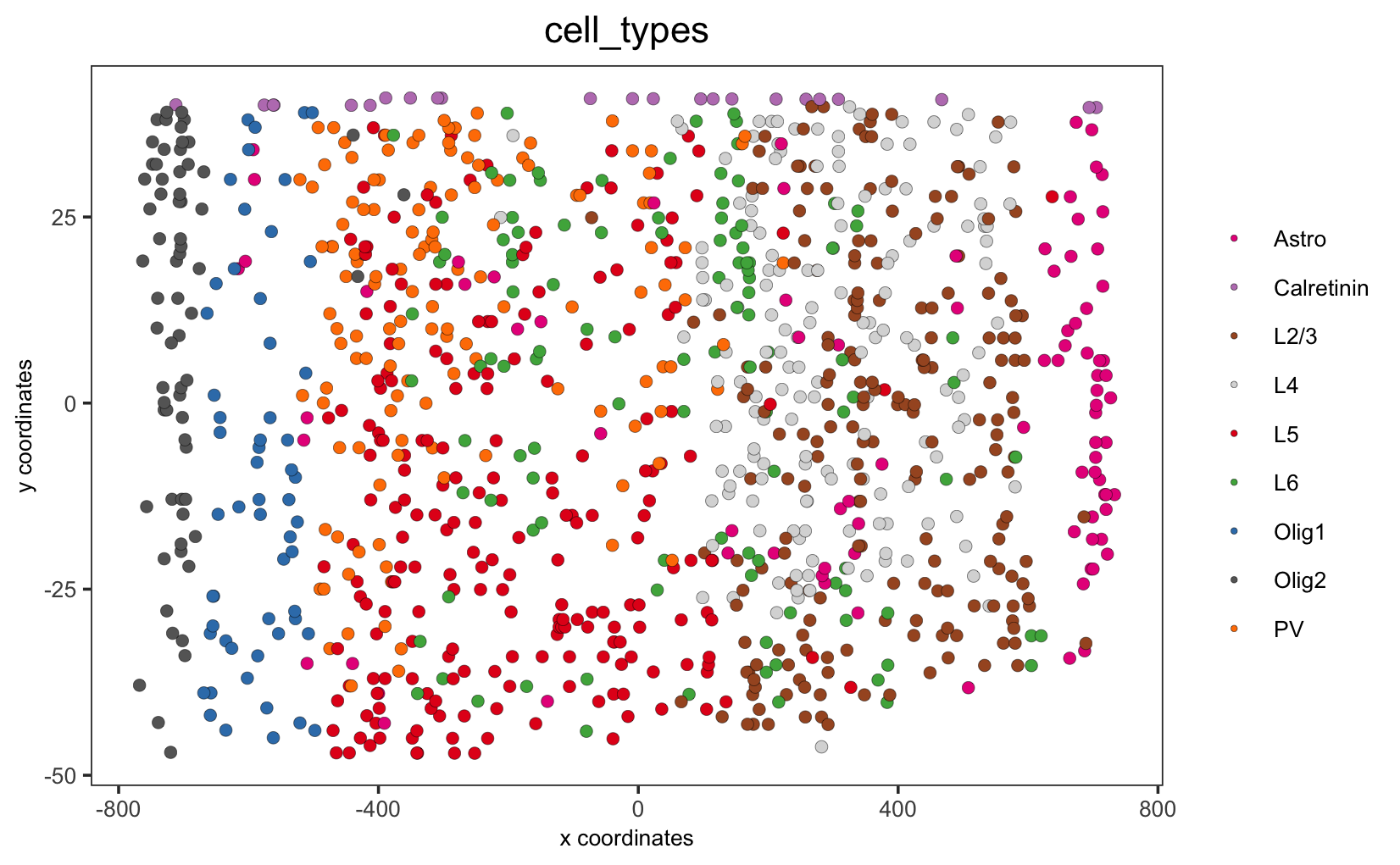
crossSectionPlot(STAR_test,
point_size = 2, point_shape = "border",
cell_color = "cell_types",cell_color_code = mycolorcode,
save_param = list(save_name = '10_a_crossSectionPlot'))
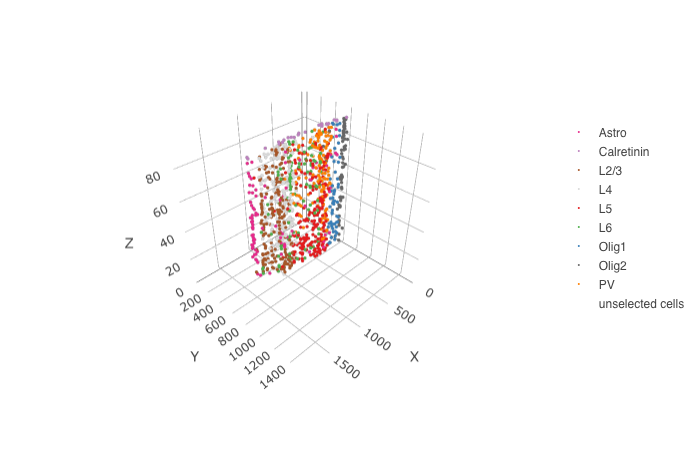
crossSectionPlot3D(STAR_test,
point_size = 2, cell_color = "cell_types",
cell_color_code = mycolorcode,axis_scale = "cube",
save_param = list(save_name = '10_b_crossSectionPlot3D'))
crossSectionGenePlot(STAR_test,
genes = "Slc17a7",
point_size = 2,point_shape = "border",
cow_n_col = 1.5,
expression_values = 'scaled',
save_param = list(save_name = '10_c_crossSectionGenePlot'))