Calculate HVG¶
-
calculateHVG()
Compute highly variable genes.
calculateHVG(
gobject,
expression_values = c("normalized", "scaled", "custom"),
method = c("cov_groups", "cov_loess"),
reverse_log_scale = FALSE,
*logbase = 2,
expression_threshold = 0,
nr_expression_groups = 20,
zscore_threshold = 1.5,
**HVGname = "hvg",
difference_in_cov = 0.1,
show_plot = NA,
return_plot = NA,
save_plot = NA,
save_param = list(),
default_save_name = "HVGplot",
return_gobject = TRUE
)
Arguments¶
gobject |
giotto object |
expression_values |
expression values to use |
method |
method to calculate highly variable genes |
reverse_log_scale |
reverse log-scale of expression values (default = FALSE) |
logbase |
if reverse_log_scale is TRUE, which log base was used? |
expression_threshold |
expression threshold to consider a gene detected |
nr_expression_groups |
number of expression groups for cov_groups |
zscore_threshold |
zscore to select hvg for cov_groups |
HVGname |
name for highly variable genes in cell metadata |
difference_in_cov |
minimum difference in coefficient of variance required |
show_plot |
show plot |
return_plot |
return ggplot object |
save_plot |
directly save the plot [boolean] |
save_param |
list of saving parameters from all_plots_save_function |
default_save_name |
default save name for saving, don’t change, change save_name in save_param |
return_gobject |
boolean: return giotto object (default = TRUE) |
Details¶
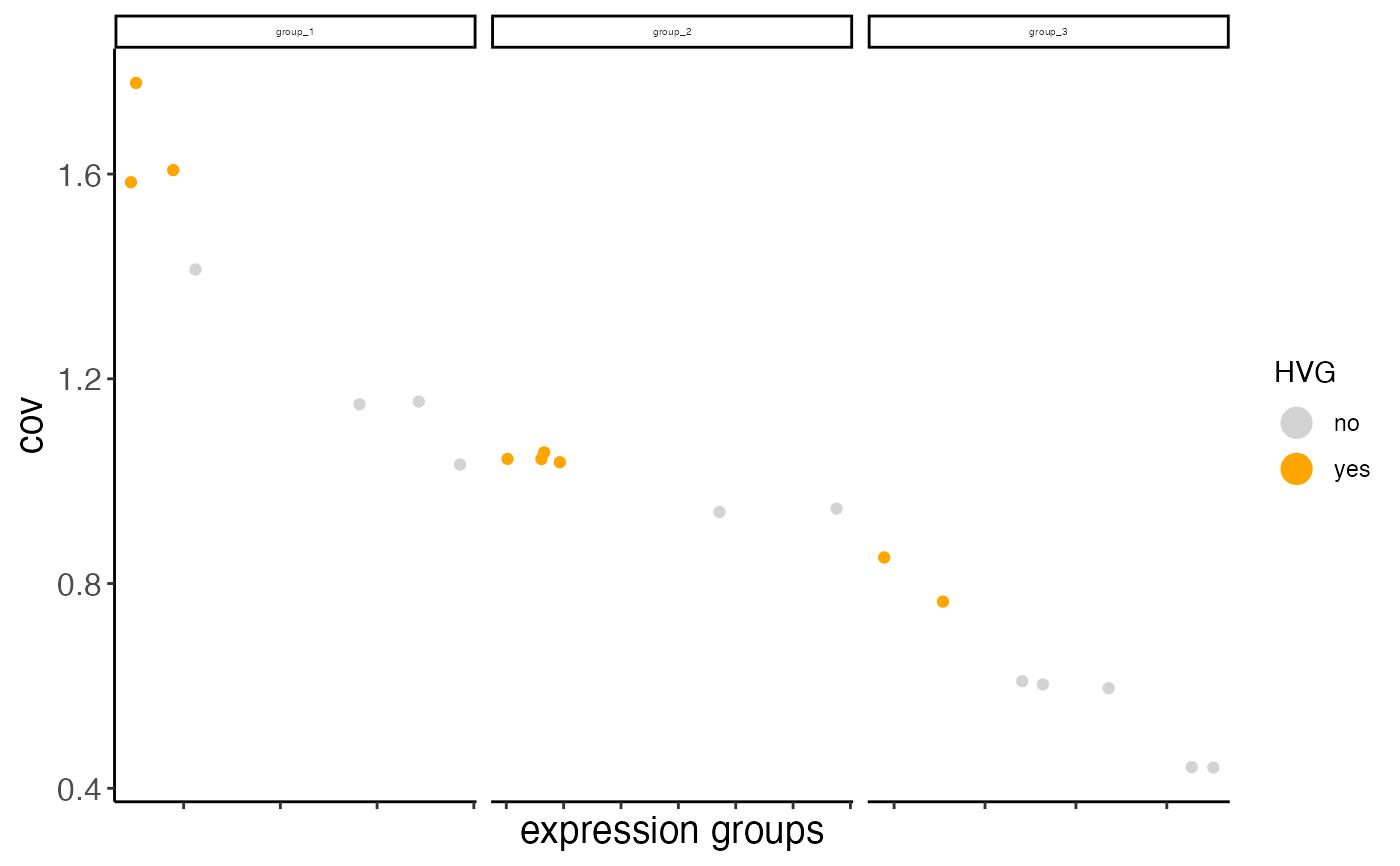
Currently we provide 2 ways to calculate highly variable genes:
1. High coeff of variance (COV) within groups:
First genes are binned (nr_expression_groups) into average expression groups and the COV for each gene is converted into a z-score within each bin. Genes with a z-score higher than the threshold (zscore_threshold) are considered highly variable.
2. High COV based on loess regression prediction:
A predicted COV is calculated for each gene using loess regression (COV~log(mean expression)) Genes that show a higher than predicted COV (difference_in_cov) are considered highly variable.
Examples¶
data(mini_giotto_single_cell) # loads existing Giotto object
# update a giotto object
mini_giotto_single_cell <- calculateHVG(gobject = mini_giotto_single_cell,
zscore_threshold = 0.1,
nr_expression_groups = 3)
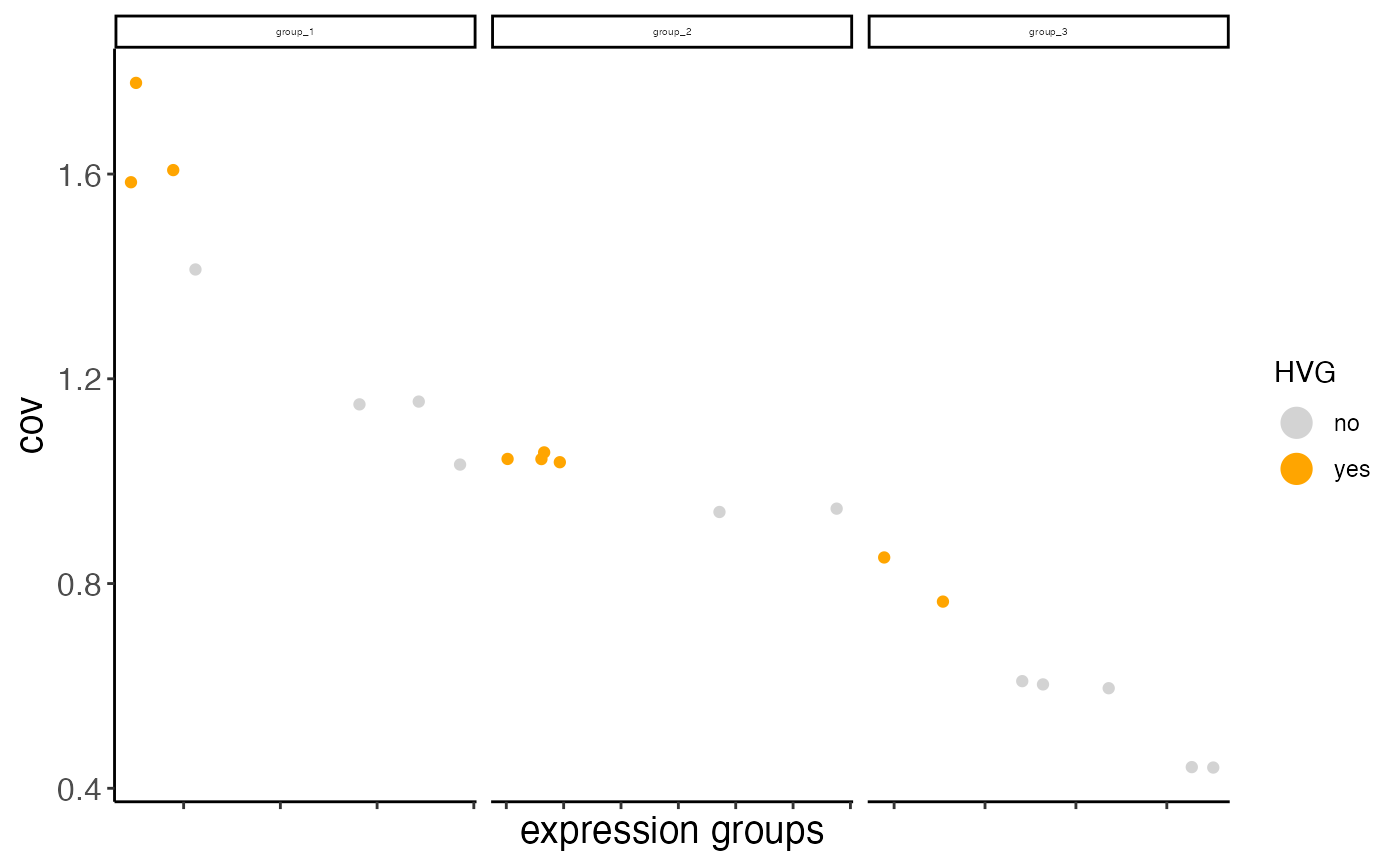
#> return_plot = TRUE and return_gobject = TRUE
#>
#> plot will not be returned to object, but can still be saved with save_plot = TRUE or manually
#>
#> hvg has already been used, will be overwritten
# return a data.table with the high variable genes annotated
hvg_dt <- calculateHVG(gobject = mini_giotto_single_cell,
zscore_threshold = 0.1, nr_expression_groups = 3,
return_plot = FALSE, return_gobject = FALSE)
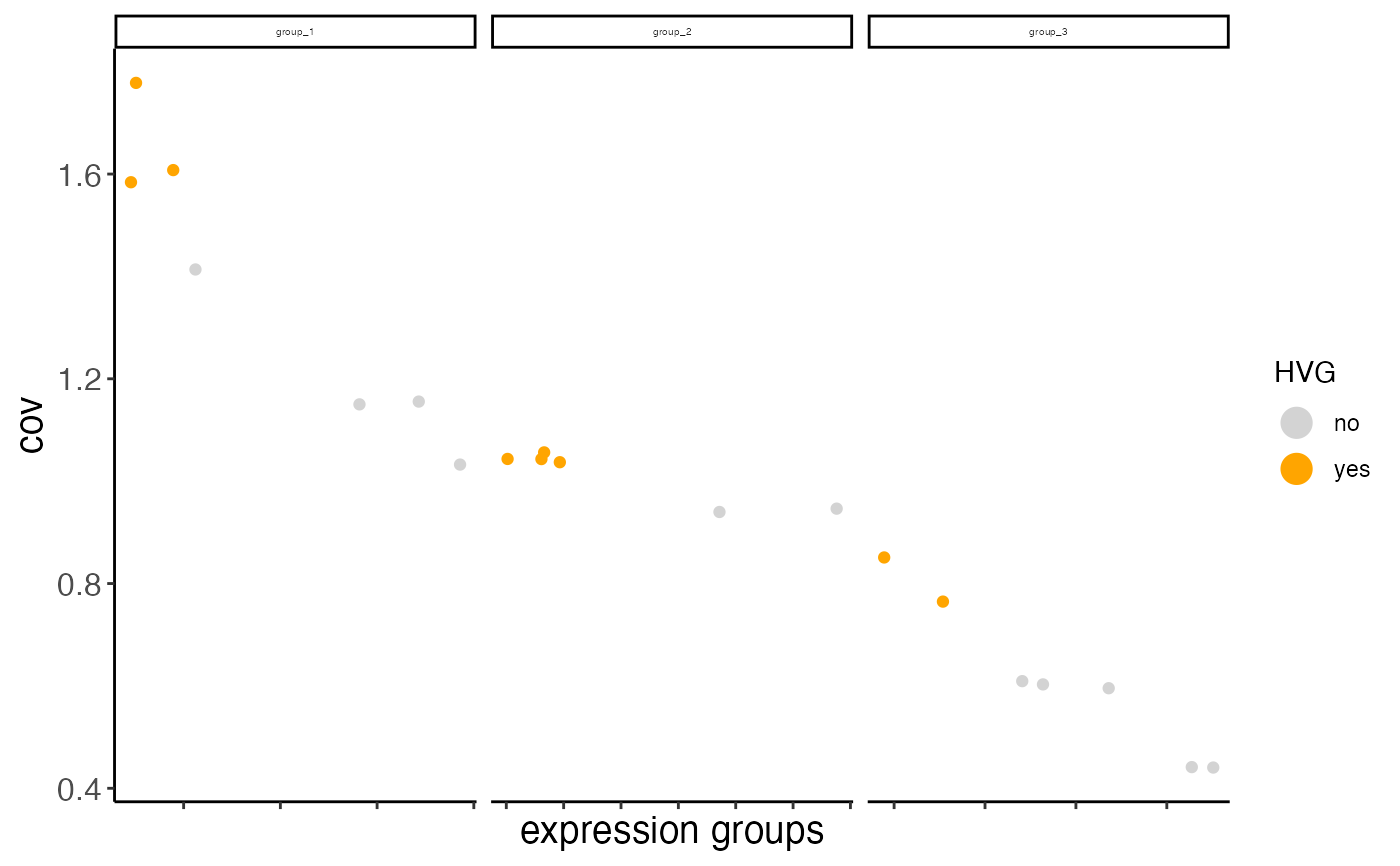
# return the ggplot object
hvg_plot <- calculateHVG(gobject = mini_giotto_single_cell,
zscore_threshold = 0.1, nr_expression_groups = 3,
return_plot = TRUE, return_gobject = FALSE)